closed
Lucia Sacchetti CEINGE-Biotecnologie Avanzate, Naples, Italy
2-years Project
Celiac disease
Area: Inflammation
- Grant: FC 007/2014
- Title: Study of the gut microbiome in the adult celiac disease pathogenesis
- Topic: Genetic and Immunology of Celiac Disease
- Duration: Biennial Project
- Principal Investigator: Lucia Sacchetti, CEINGE-Biotecnologie Avanzate, Naples, Italy
- Partnerships: Giovanna Del Vecchio Blanco, Department of System Medicine (Gastroenterology Unit), University of Rome Tor Vergata, Italy; Paola Salvatore, Department of Molecular Medicine and Medical Biotechnologies, University of Naples Federico II, Italy
Publications originated from the Project:
- V. D’Argenio et al. Metagenomics Reveals Dysbiosis and a potentially pathogenic N. flavescens strain in duodenum of adults celiac patients. The American Journal of Gastroenterology (2016); doi:10.1038/ajg.2016.95 https://pubmed.ncbi.nlm.nih.gov/27045926/
- V. D’Argenio et al. No change in the mucosal gut mycobioma is associated with celiac disease-specific microbiome alteration in adult patients. The American Journal of Gastroenterology (2016); doi:10.1038/ajg.2016.227 https://pubmed.ncbi.nlm.nih.gov/27808136/
- Iaffaldano L, Granata I, Pagliuca C, et al. Oropharyngeal microbiome evaluation highlights Neisseria abundance in active celiac patients. Sci Rep. 2018;8(1):11047. Published 2018 Jul 23. doi:10.1038/s41598-018-29443-1 https://pubmed.ncbi.nlm.nih.gov/30038321/
- Labruna G, Nanayakkara M, Pagliuca C, et al. Celiac disease-associated Neisseria flavescens decreases mitochondrial respiration in CaCo-2 epithelial cells: Impact of Lactobacillus paracasei CBA L74 on bacterial-induced cellular imbalance. Cell Microbiol. 2019;21(8):e13035. doi:10.1111/cmi.13035 https://pubmed.ncbi.nlm.nih.gov/31042331/
THE STUDY
Project rationale and aims
We hypothesize that alterations of the gut microbiome could concur in compromising epithelial function in adult celiac disease (CD) patients and that they could be another trigger of the disease besides gluten and host genetics.
Aim 1. To use metagenomic technologies (NGS) to characterize the duodenal microbiome of adult control subjects and active and non active CD patients. A comparison between the three groups will reveal the microbiome profiles associated with the active and non active stage of the disease, and qualitative and/or quantitative differences in duodenal microbial composition (up to genus level) probably related to CD pathogenesis.
Aim 2. To fully characterize, using a culture-based approach, all the gut cultivable bacteria, and, after comparing these microbiological data with the metagenomic data, to isolate bacteria that differ significantly between CD patients and controls.
Aim 3. To test in a polarized epithelial cell model: (a) whether selected CD-altered
bacteria modulate inflammation, and (b) the effect of selected CD-altered bacteria in influencing intestinal epithelial functions. In particular, we will evaluate invasiveness and the effect on intracellular trafficking.
Research plan and results obtained
Aim 1. A) Proteobacteria was the most abundant whereas the Actinobacteria and Firmicutes phyla were the less abundant phyla in the microbiome profiles of active CD patients than in the other 2 groups. Members of the Neisseria genus, within the Betaproteobacteria class were significantly (p=0.03) more abundant in active CD patients than in the other 2 study groups. B) As fungi also contribute to gut microbiota, we studied the duodenal mycobiome composition in the same cohort of individuals without finding any CD-associated fungal alteration. C) We characterized by NGS in another cohort of active CD patients, GFD CD patients and control subjects, also oropharyngeal microbiome, to test the hyphotesis of a continuum from the oral niche to the duodenum in microbiome of CD patients. Interestingly, a similar core microbiome with significant higher levels of the Neisseria genus was detected in the oropharynx of active CD than in the other two groups.
Aim 2. Standard microbiological cultures of duodenal and oropharyngeal specimens revealed a cultivable aerobic biodiversity in duodenal microflora of the samples tested. In particular, Neisseria flavescens (Nf) was viable in all oropharyngeal samples and in duodenal mucosa samples from active CD patients (CD-Nf), but was undetectable in the duodenum from controls.
Aim 3. A) We investigated in vitro the pro-inflammatory properties of the lysates of Nf strains isolated from active CD subjects using both murine DCs and human monocyte-derived DCs and found that the production of both TNF-alpha and IL12p40 was higher in Nf -stimulated DCs than in either untreated or LPS treated DCs (p<0.0001). Latter results indicate that Nf strains imprinted a pro-inflammatory phenotype on both murine and human DCs in vitro. Further, we found increased epithelial expression of the inflammation markers HLA-DR and COX-2 in ex-vivo mucosal explants from control subjects when exposed to CD-Nf bacteria for up to 6 hours. B) By using confocal microscopy we compared the intracellular localization of CD-Nf with that of control-Nf up to 6 hours after infection in CaCo-2 epithelial cells. This experiment showed that while CD control-Nf clearly colocalized with LAMP1 (marker of the late/endosome-lysosomal pathway) most CD-Nf escaped this intracellular compartment.
Experimental Design and Methodologies
Study groups. Two cohorts were studied, the first consisted of 41 unrelated Caucasian individuals (20 active CD patients, 15 control subjects and 6 GFD) and was investigated for duodenal microbiome characterization, whereas the second consisted of 56 Caucasian individuals (14 active CD patients, 20 control subjects and 22 GFD) and was investigated for oropharyngeal microbiome characterization.
Methods. Total DNA was extracted by using QIAamp DNA Mini Kit (Qiagen,Venlo, Netherlands). NGS reactions were carried out on a 454 Genome Sequencer instrument (Roche, Penzberg, Germany)(bacterial duodenal microbiome and mycobiome) or on IlluminaMiSeq System (Illumina, San Diego, CA) (oropharyngeal microbiome). Sequences were analyzed using QIIME v. 1.9.1. Culture-dependent microbiological analysis, isolation and identification (by mass spectrometry) of bacterial species were also performed. Splenic and human bone marrow-derived dendritic cells (DCs) (isolated from 6 C57BL/6 mice and 3 healthy volunteers, respectively) and ex-vivo mucosal explants from 3 control subjects were used for testing the inflammatory power of CD-Nf in in vitro experiments. CaCo-2 epithelial cells were obtained from the Bank of Human and Animal Continuous Cell Lines (CEINGE-BiotecnologieAvanzate) and used for cellular internalization experiments of CD-Nf.
Potential pitfalls and caveats
We did not encounter any difficulties in the project.
Conclusions and Discussion
CD-associated duodenal dysbiosis is not yet clearly defined and the mechanisms by which gut CD-associated dysbiosis could concur to CD development or exacerbation remains largely unknown. In the attempt to shed light on this issue, we used 16S rRNAmetagenomics to determine whether the duodenum microbiome differed among 3 groups of adult individuals: active CD patients, GFD CD patients and controls. We found that members of the phylum Proteobacteria were more abundant, and those of the Firmicutes and Actinobacteria phyla less abundant in active CD than in either GFD patients or controls (Figure 1).
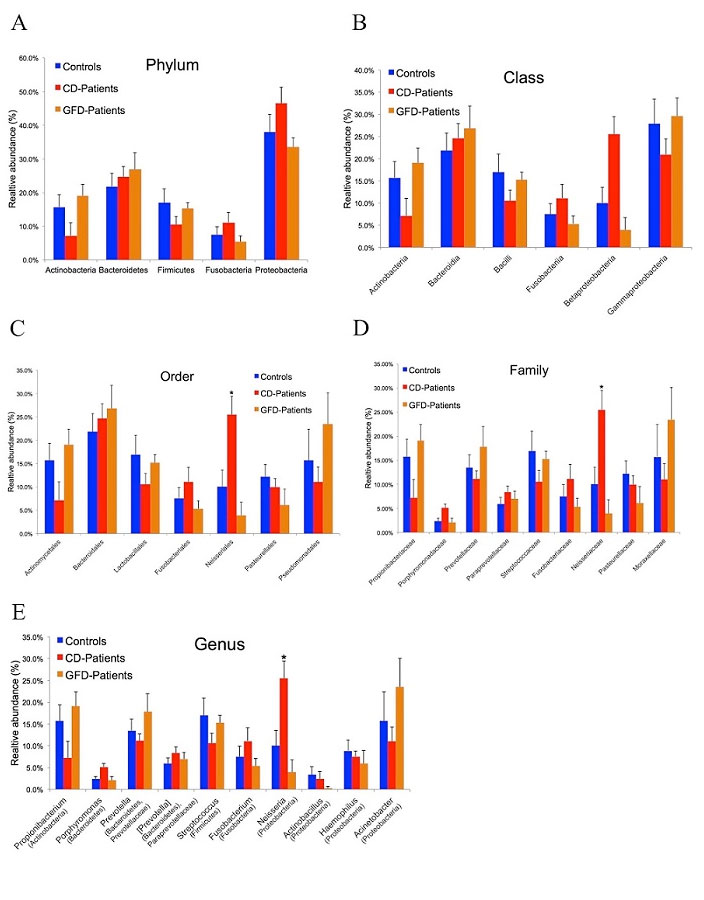
Figure 1 – Duodenal microbiome taxonomic composition in controls, active and GFD CD patients. (A) Relative abundance at phylum level. Proteobacteria was the most abundant phylum in all 3 groups with an average abundance of 39.3%. (B) Class level classification in the 3 groups showed a decreasing trend for the Betaproteobacteria class in GFD patients (4%) and controls (10%) versus CD patients (26%). The Gammaproteobacteria class was less abundant in CD patients (21%) than in controls (28%) and GFD patients (30%). (C) Order level classification revealed a significant difference (p=0.009) in the Neisseriales order among controls (~10%), GFD patients (~4%) and CD patients (~26%). (D) Also at family level, the Neisseriaceae family was significantly less abundant (p=0.01) in controls (~10%) and GFD patients (4%) than in CD patients (~32%). (E) At genus level, Neisseria was significantly more abundant (p=0.03) in active CD patients (26%) than in controls (10%) and GFD patients (4%). Taxa (in parentheses) refer to the phylum to which the genus belongs. The data reported are filtered by a frequency higher than 1%. Error bars indicate standard error. Asterisks refer to taxa that differed significantly among the 3 groups (p<0.05, ANOVA). From V. D’Argenio et al. Am. J. gastroenterol. 2016
A similar dysbiosis was previously reported in the gut microbiome of adult CD patients with gastrointestinal symptoms (Wacklin P. et al. Inflamm. Bowel Dis, 2013), confirmed in the gut microbiome of GFD CD patients with persistent symptoms (Wacklin P. et al. AJG, 2014) and in biopsy specimens from active CD children when compared to GFD and controls (Sànchez E. et al. Appl. Environ.Microbiol, 2013). All the above data support the concept that the gut dysbiosis associated with the active stage of CD is characterized by an increase of Proteobacteria and a decrease of Firmicutes phyla. However, no other group has previously explored how specific bacterial species detected in the duodenal mucosa of CD patients could contribute to the CD-associated gut malfunction. Within the bacterial genera detected in the gut microbiome of the 3 study groups the relative abundance of the Neisseria genus was significantly higher in active CD patients (26%), than in either GFD patients (4%) or controls (10%) and we identified N. flavescens as the major species present in the CD gut microbiome among the Neisseria isolates. We explored the inflammatory effect of CD-Nf, because overgrowth of pathogenic bacteria could lead to aberrant activation of the immune system. Interestingly, we found that Nf may promote a pro-inflammatory phenotype in both murine and human DCs characterized by high levels of inflammatory cytokines (IL-12p40 and TNF-α) (Figure 2).
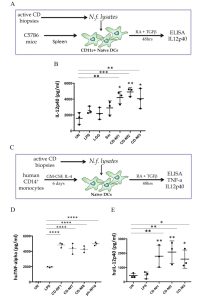
Figure 2 – N. flavescens isolated from CD patients induce a pro-inflammatory phenotype on dendritic cells. Filtered bacterial lysates from Neisseria flavescens (Nf), isolated from 5 active celiac patients (CD-Nf1, 2, 3, 7 and 8), promoted the production of pro-inflammatory cytokines by dendritic cells (DCs). CD11c+ DCs isolated from the spleens of C57B6 mice were cultured for 48h in the presence of retinoic acid (RA) and TGF-beta? with filtered Nf lysates from three active CD patients (A). All three tested bacterial lysates promoted the production of IL-12p40 by murine DCs, whereas untreated (stars upon horizontal bars), LPS-treated (stars) and LGG-treated DCs did not (B). Human monocyte-derived DCs were cultured for 48h in the presence of RA and TGF-beta? with filtered Nf lysates isolated from active CD patients (C). TNF-a levels were higher in supernatants of human DCs treated with Nf1 Nf7 and Nf8 bacterial strains isolated from small intestinal biopsies of CD patients, and in those challenged with Nf10 isolated from a pharyngeal swab of a control subject (D), than in with bacterial LPS, used as a pro-inflammatory positive control (stars upon horizontal bars) and in untreated (UN) cells (p<0.0001 for each of the treatments). IL-12p40 levels were higher in supernatants of human DCs challenged with Nf1,2,3 filtered lysates than in with bacterial LPS (stars) used as a pro-inflammatory positive control, and in untreated (UN) cells (stars upon horizontal bars) (E).
Dot plots represent values from 3 independent experiments. Each experiment was performed in duplicate. A 2-way ANOVA was performed to check for inter-group differences.
* p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. From V. D’Argenio et al. Am. J. gastroenterol. 2016
Furthermore, IFN-gamma?, TNF-α and IL-23 levels were higher in the culture supernatants of treated with CD-Nf than in untreated biopsies from controls. These data support our hypothesis that Nf contributes to promoting inflammation in the small intestine. Whether this inflammation precedes or follows the onset of full-blown CD remains, however, to be studied. It is feasible that the inflammatory conditions occurring in the gut of CD patients may favor the colonization of Nf strains thus promoting the maintenance of a pro-inflammatory immune response; on the other hand, under certain conditions (environmental factors, viral infections, genetic predisposing factors, etc.), Nf colonization of the duodenal tract is favored, and this event could contribute to triggering CD development in genetically susceptible individuals. At present, the induction of inflammatory responses in DCs and in ex-vivo control mucosal explant supports the first possibility. Interestingly, oropharyngeal and duodenal microbiomes in active CD patients were comparable, being characterized by an abundance of Nf within the most abundant Proteobacteria phylum, consistent with the hypothesis of a continuum from the oral niche to the duodenum in active CD patient. Conversely, the oropharyngeal microbiome in GFD patients matched the one of the controls.
In conclusion, marked dysbiosis and an abundance of a peculiar CD-Nf strain characterize the duodenal and oropharyngeal microbiomes of active CD patients, which suggests that the CD-associated microbiota could contribute to the many inflammatory signals in this disorder. Further, given the control-like oropharyngeal microbiome after GFD, we may speculate that microbiome characterization in the oropharynx could be used in monitoring the efficacy of GFD.